
Medical Device Documentation: Start Early, Stay Iterative
The Critical Role of Documentation in Medical Device Development:
The process of bringing a medical device to market can be complex, but it doesn’t have to be daunting. One of the most important things to understand is that the time to think about documentation is at the beginning—not at the end. While it may seem like a chore to document everything from the start, doing so early on ensures that you won’t be scrambling at the last minute.
Documentation should be iterative, a process that evolves alongside the product development cycle. By integrating documentation and risk management from the outset, you’ll save time, reduce risks, and ensure smoother regulatory submissions later. It’s about building on what you know, refining your processes, and ensuring that everything is traceable and compliant as you move forward.
⚠️ Critical Note: We always suggest starting with a Quality Management System (QMS) and partnering with a regulation expert or an experienced product manager to lay the proper groundwork. This ensures that the medical device development process remains compliant and efficient from the very beginning.
Key Documentation and Risk Management Requirements for Medical Device Compliance
When developing a medical device, there’s a handful of essential documents you need to keep in check to stay on the right side of regulations. Think of it as packing for a trip—you don’t want to forget the essentials! Key standards like ISO 14971 guide you through managing risks, IEC 62304 covers the software development lifecycle, ISO 17664 ensures proper reprocessing of reusable devices, and ISO 11607 sets the requirements for packaging sterile medical devices. Get these documents right, and you’ll have everything you need neatly packed and ready for a smooth journey through compliance.
1. Device Description, Labeling, Instructions for Use, and Packaging
This stage is not usually the most exciting part for engineers, but if incorporated into the development process iteratively, it can effectively generate most of the necessary documentation. If it feels like a chore, it’s often because it’s being done too late—meaning there may be many design loose ends that need to be tightened up, which makes the process more complicated and frustrating.
At the heart of your documentation package, you’ll need to include:
Device Description: Clearly outline how your product works and its intended purpose. This helps everyone involved—from regulators to users—understand exactly what the device is and what it does.
Labeling and Instructions for Use: Provide detailed information on how to safely use the device, including any risks or safety precautions. Clear labeling is essential for preventing misuse and ensuring patient safety.
Sterilization and Packaging Information: This is a critical part that ensures your device stays safe and effective from the factory to the point of use. Packaging isn’t just about keeping the device clean—it must protect the device from physical damage, contamination, and environmental factors like moisture or air that could compromise sterility or performance.
Medical device packaging must comply with recognized standards such as ISO 11607, which specifies how to design, test, and validate packaging that keeps sterile devices safe until use. This includes both the primary packaging (the immediate container around the device) and secondary packaging (additional layers like boxes). Most people tend to focus only on testing the device itself, but it’s important to remember that the packaging also has to pass rigorous testing. After all, if the packaging fails, it can compromise the sterility and safety of the device inside—so packaging is just as critical to get right.
Packaging Testing: To meet these standards, your packaging must go through rigorous testing, which includes:
Integrity Testing: Ensuring the package doesn’t leak, tear, or otherwise fail during transportation and storage.
Sterility Maintenance: Confirming that sterilization is preserved and intact until the package is opened.
Simulation of Real-World Conditions: Testing packaging under stresses such as drops, vibrations, temperature changes, and humidity to ensure it holds up in the environments it may face before use.
Early preparation and inclusion of this information in your documentation package help avoid confusion or delays later in the regulatory submission process. It demonstrates that you’ve thoughtfully designed your device and packaging to protect patient safety and device effectiveness throughout the product’s entire lifecycle.
2. ISO 14971 and IMDRF’s Adverse Event Terminology: A Comprehensive Guide to Medical Device Risk Management for Founders
As a founder venturing into the medical device industry, one of your top priorities is ensuring your product is safe, effective, and compliant with regulatory standards. Achieving this requires a robust risk management process that doesn’t stop once your device leaves the development lab—it must span the entire lifecycle of your product. Two key frameworks will guide you through this: ISO 14971, the global standard for risk management, and the International Medical Device Regulators Forum’s (IMDRF) Adverse Event Terminology (AET) system, which supports ongoing safety monitoring post-market.
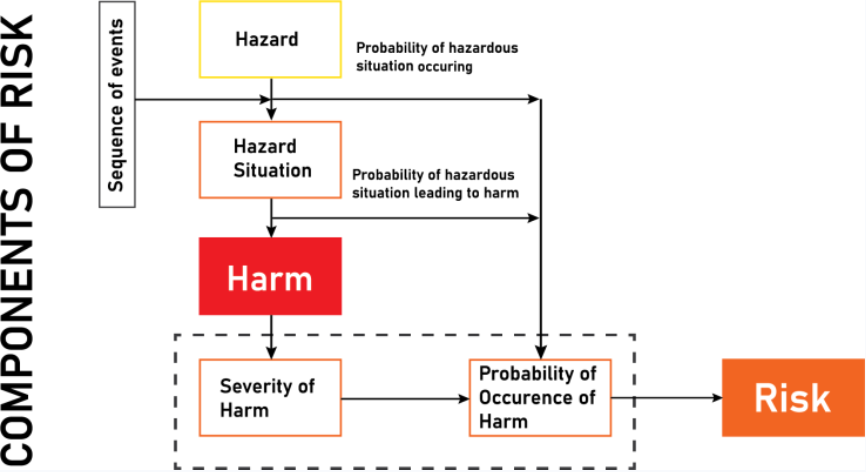
ISO 14971 is the cornerstone of medical device risk management. It mandates that you implement a comprehensive process to identify, evaluate, control, and monitor risks associated with your device throughout its entire lifecycle.
Risk Analysis: This is where you identify potential hazards—anything that could cause harm—early on. For example, if your device uses electricity, you must analyze potential electrical hazards during the design phase.
Risk Evaluation: After identifying hazards, you assess their likelihood and severity to decide if the risk is acceptable or if further action is needed.
Risk Control: If risks are unacceptable, you implement controls such as design changes or protective features—like adding insulation to prevent electrical shock.
Residual Risk Assessment: After applying controls, you reassess remaining risks to ensure they meet safety thresholds before market release.
Unlike narrower techniques like Failure Modes and Effects Analysis (FMEA), which focus mainly on component failures, ISO 14971 addresses a wider range of risks—covering normal use, misuse, abnormal conditions, and fault scenarios throughout the product’s lifecycle. This holistic approach is essential to meet regulatory demands by agencies such as the FDA and the European Union.
IMDRF’s Adverse Event Terminology (AET): Ensuring Safety Beyond Market Launch
But your responsibility doesn’t end when your device hits the market. As more patients and providers use your device, new risks can emerge. Managing these post-market risks requires a standardized approach to tracking and reporting adverse events—this is where IMDRF’s Adverse Event Terminology (AET) becomes invaluable.
AET is a globally accepted language for describing and classifying problems with medical devices, enabling clear, consistent communication between manufacturers, healthcare professionals, and regulators worldwide.
Why does this matter? Before AET, different countries and organizations used their own terms to describe device problems, causing confusion and delays in addressing safety issues. With AET, everyone uses the same “dictionary,” which improves:
Consistent Reporting: Whether a device malfunction, patient harm, or component failure, events are categorized the same way everywhere.
Early Detection of Trends: Regulators like the FDA or EMA can quickly spot patterns indicating emerging risks.
Swift Regulatory Response: Standardized classifications help trigger faster recalls, safety alerts, or design improvements.
Global Regulatory Alignment: Harmonized terminology supports international cooperation, streamlining compliance in multiple markets.
AET classifies adverse events hierarchically. For instance, a “device malfunction” might break down into specific issues like “battery failure” or “software glitch,” while “user outcome” categories capture patient injuries or harms. This detailed categorization applies across the product lifecycle—from clinical trials to everyday use—ensuring you maintain vigilant safety monitoring at all stages.
Why Integrate ISO 14971 and AET from the Start?
Incorporating ISO 14971’s risk management framework early in your design process—and aligning it with post-market surveillance systems like AET—creates a continuous feedback loop that enhances device safety and compliance:
Pre-Market: Design and test your device with a full understanding of potential risks, and plan how to monitor and mitigate them after launch.
Post-Market: Use AET to report adverse events in a standardized way that meets regulatory requirements and helps you quickly detect new risks or failures.
Regulatory Compliance: Both frameworks are recognized by leading regulatory agencies worldwide, so adherence reduces the risk of costly delays, market withdrawals, or legal issues.
Patient Safety: Most importantly, this integrated approach ensures patient safety is continuously prioritized, building trust in your device and brand.
3. IEC 62304: Software Lifecycle Processes
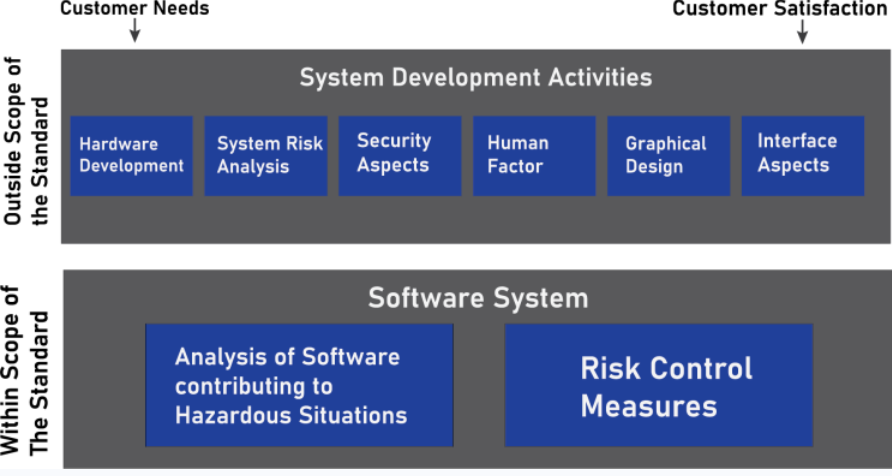
For medical devices with software components, IEC 62304 provides a framework for developing and maintaining medical software. This standard ensures the software is safe, effective, and reliable, and emphasizes the importance of documentation throughout the software lifecycle:
Software Development Life Cycle (SDLC): Documentation must include planning, design, development, testing, and maintenance. Every decision must be backed up by evidence.
Risk Management for Software: Identify risks specific to software, such as algorithm errors or crashes, and integrate them into the device’s overall risk management plan.
Software Validation and Verification (V&V): Prove that the software meets its intended use and doesn’t introduce new risks into the device.
By adopting IEC 62304, you’ll ensure that your software components are rigorously developed, thoroughly tested, and validated—all backed by strong documentation. Again, this process should begin early in development and evolve throughout.
4. ISO 17664: Reprocessing of Medical Devices
For reusable medical devices, ISO 17664 is critical. This standard specifies the reprocessing procedures that must be documented, including cleaning, disinfection, and sterilization steps.
Reprocessing Instructions: The manufacturer must provide clear, validated instructions for how to properly reprocess the device between uses, ensuring that it can be safely reused without compromising its performance.
Validation: All reprocessing methods must be validated to ensure that the device remains safe for use after multiple cycles of cleaning and sterilization.
Risk Management for Reprocessing: Assess risks associated with improper cleaning, sterilization failure, or contamination during reprocessing, and take corrective actions to mitigate those risks.
Maintaining detailed reprocessing instructions and conducting risk assessments for this phase ensures that devices stay safe and effective, even after multiple uses.
Continuous Risk Management and Documentation
Documentation and risk management don’t end once your device is on the market. Post-market surveillance and continuous risk assessment are critical for maintaining compliance and safety. As your device is used in real-world settings, new risks may emerge. By keeping your risk management file updated with production data, complaints, and post-market surveillance, you ensure that the device remains safe for its users.
Ongoing Feedback Loop
Once your device is in the market, production and post-production activities provide valuable feedback. This can be used to:
Update the risk analysis as new risks emerge.
Refine risk control measures to ensure safety.
Ensure that the reprocessing steps for reusable devices remain effective.
This iterative loop is crucial to continuously monitor the safety and effectiveness of your device, and ensure that any identified risks are managed appropriately throughout the product’s lifecycle.
Conclusion
To wrap up, managing documents well is key to developing successful medical technology products. It’s not just about keeping things organized—proper document management helps your team work together smoothly and keeps track of every step along the way. This is especially important because medical devices must meet strict regulations to ensure they are safe and effective. Standards like ISO 13485, which focuses on quality management, and ISO 14971, which deals with identifying and managing risks, guide how documents should be handled during product development. Using a good document management system that follows these standards makes it easier to pass audits, reduces risks, and speeds up getting your product to market. In short, thoughtful document management protects your company and helps build better, safer medical devices.
Ready to bring your product to life?
Book a consultation with Jeremiah Landi and explore how our turnkey engineering can support your next breakthrough. Follow our newsletters and get resources for building better products.