
Demystifying Regulatory Pathways for Medical Devices
Navigating the regulatory landscape is one of the biggest challenges in developing a medical device. Too often, companies begin engineering a product without considering the regulatory impact of both what they are designing and what they are not.
Just like with IP protection, it’s critical to define what matters and what doesn’t before involving regulatory bodies. Once the process begins, reversing course becomes difficult and costly. We’ve seen many products make this mistake—the goal with regulation isn’t to aim for a higher classification but to achieve the lowest viable one, reducing both risk and compliance costs.
To help simplify this process, we’re launching a newsletter featuring long-form articles designed to guide both newcomers and industry veterans. For those new to the field, these articles will break down key considerations and common pitfalls. For seasoned companies, they’ll serve as a valuable resource for refining and optimizing regulatory strategies.
Up on deck for the coming weeks and months:
Ensuring Safety & Compliance: Medical Device Sterilization
Anodization in a Sterile World: Choosing the Right Process for Medical Applications
Selecting the Right Silicone: Considerations for Medical Device Innovation
Surgical Device Failure Modes: Identifying Risks and Enhancing Patient Safety
FDA Regulatory Pathways Overview
Before bringing a medical device to market in the U.S., obtaining FDA clearance or approval is a crucial requirement—yet it’s often overlooked during early fundraising. Neglecting this step can create significant barriers to securing Letters of Intent (LOIs), ultimately leading to fundraising challenges.
This is a critical pain point for startups. Failing to secure funding is one of the most common breaking points for early-stage companies, not only disrupting product delivery but also causing a loss of momentum that can jeopardize hard-earned relationships with doctors and hospitals—relationships that often take years to build.
The FDA uses different pathways based on the device's classification. For instance, a toothbrush will not undergo the same scrutiny as a pacemaker. Understanding your device’s classification is vital as it influences both product design and regulatory requirements. Engaging a regulatory consultant or attorney early can save significant time and costs.
Example:
Years ago, we worked on a device that “sterilized” pens. Initially, the product was classified as a medical device, requiring full FDA approval; however, after consulting regulatory attorneys, we redefined the function from “sterilize” to "disinfectant." This redefinition allowed us to bypass the lengthy approval process within the FDA.
Medical Device Classifications
The FDA categorizes medical devices into three main classes based on risk and regulatory requirements:
Class I: Low risk (e.g., bandages), with most devices exempt from premarket notification.
Class II: Moderate risk (e.g., X-ray machines), often requiring 510(k) clearance.
Class III: High risk (e.g., pacemakers), requiring Premarket Approval (PMA).
A fundamental rule we follow: the higher the classification, the more costly and rigorous the approval process becomes. To put this in perspective, 47% of approved devices are Class I, while only 10% fall under Class III. Understanding your device’s classification early can significantly streamline the regulatory pathway and reduce unnecessary hurdles.
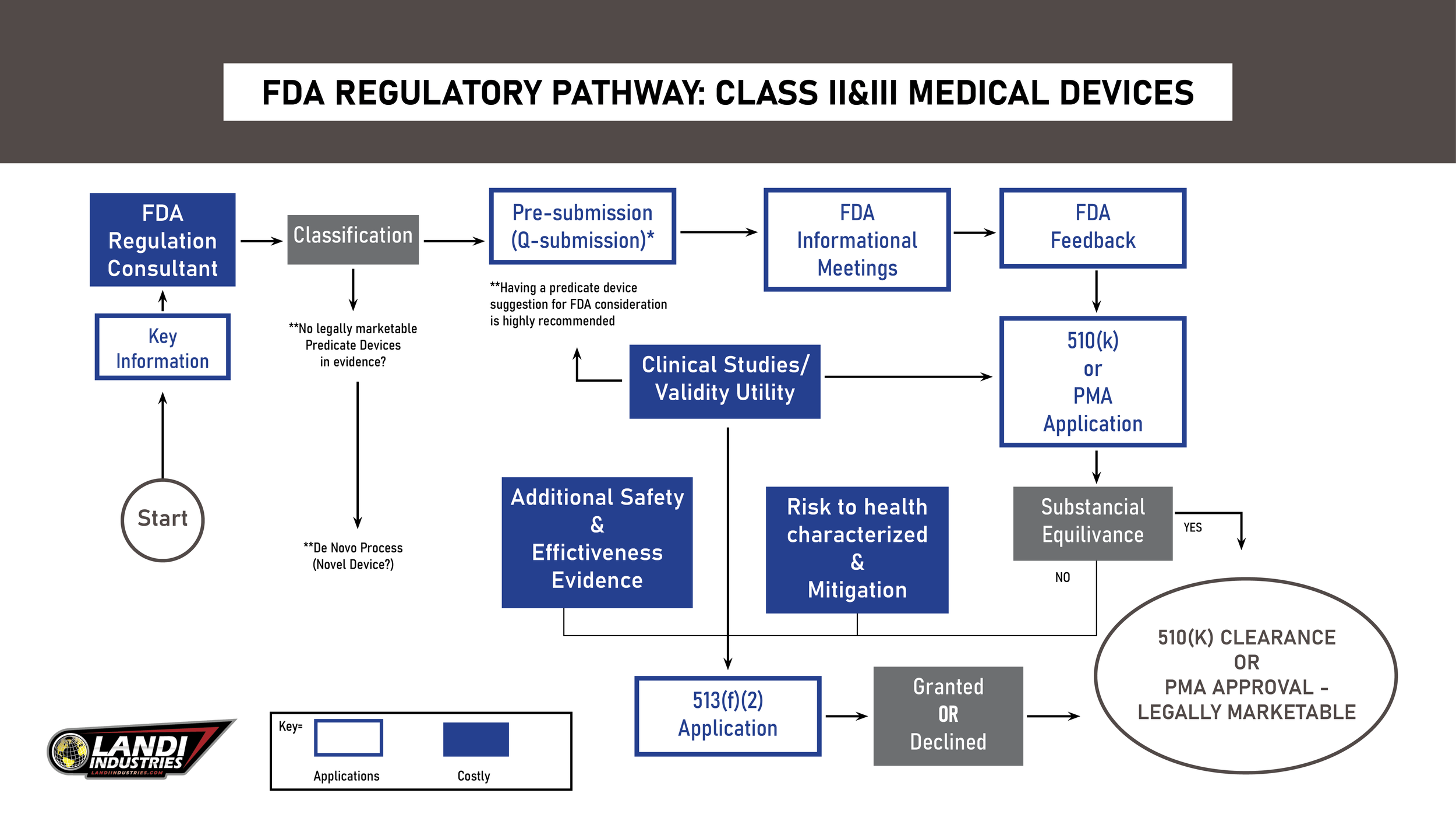
What is a 510(k)?
The 510(k), or Premarket Notification, is the most common regulatory pathway for medical devices seeking approval to enter the U.S. market. It primarily requires two key elements:
Predicate Device: A similar product already on the market.
Substantial Equivalence: The new device must share the same intended use and comparable technological characteristics as the predicate.
At the core of the 510(k) process is the concept of substantial equivalence. Essentially, the goal is to demonstrate to the FDA that the device you intend to bring to market is broadly similar to an existing device already on the market (the predicate). The logic behind this submission is straightforward: if the predicate device is already deemed safe and effective, then the new device, being similar, should also be safe and effective.
The FDA reviews 510(k) submissions within 90 days. If the device is found to be substantially equivalent, it will receive the same classification as its predicate. If not, it may require a more stringent Premarket Approval (PMA) process.
When preparing your 510(k) submission, it is crucial to ensure the following elements are clearly presented:
Demonstrate that your medical device is ‘substantially equivalent’ to a predicate device already on the market.
Provide detailed technical, safety, and performance data to substantiate that your device is safe and effective.
Prove that you have a robust quality management system and a comprehensive risk management process in place.
This is why it’s vital to engage a company that specializes in regulatory compliance before initiating the development work. Having regulatory experts involved early simplifies the process and ensures that all necessary tests and documentation are in place. As I often say, the job of building these devices is what bioengineers do best, but navigating the regulatory requirements is not part of the engineering process—it’s a specialized task that requires careful attention.
**as a footnote, we do a lot of work in the ClassIII arena (eg. Pacemakers, defibrillators, and digital mammography systems) and will save some of the more complex nuances for future articles.
Exemptions
To briefly touch on a broader topic, certain devices are exempt from FDA premarket notification or approval requirements. For example:
Class I Devices: Many are automatically exempt from premarket notification.
Class II Devices: Some devices are exempt but still require FDA registration.
Additionally, there are other pathways worth mentioning, though they are less common. The Investigational Device Exemption (IDE) applies to experimental devices, while the Humanitarian Device Exemption (HDE) applies to devices intended to benefit small patient populations.
While these exemptions exist, they are not something we encounter frequently. However, no FDA article would be complete without a brief mention of them.
Pro Tip: Leverage FDA Resources
The FDA provides detailed guidelines and resources on its website, of which most of this was condensed from. Exploring these resources can clarify processes and requirements for your specific device.
In Summary
Getting involved in the medical device community—such as organizations like SOPE, HIMSS, Life Sciences—can provide invaluable support. Surrounding yourself with other founders in the industry helps clarify the complexities of the regulatory process and can expedite finding trusted consulting firms when needed.
However, the most crucial factor is understanding the regulatory pathways early on. This proactive approach not only helps avoid costly mistakes but also streamlines the journey, bringing your medical device to market more efficiently.